Beta-thalassemia là bệnh di truyền phổ biến ở Việt Nam. Trong trường hợp người mắc bệnh beta-thalassemia ở thể nặng hoặc thể trung gian cần được chăm sóc y tế cả đời, điều này là gánh nặng cho bản thân, gia đình và xã hội. Vì vậy việc dự phòng để hạn chế sinh ra các thể bệnh nặng và trung gian là vô cùng quan trọng.
Bệnh tan máu bẩm sinh thể Beta (Beta thalassemia) là gì?
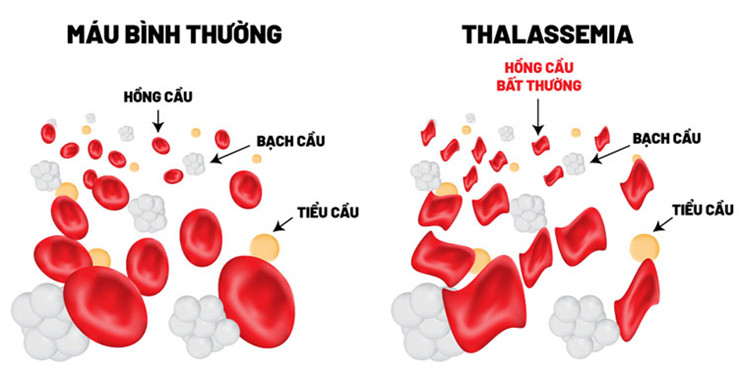
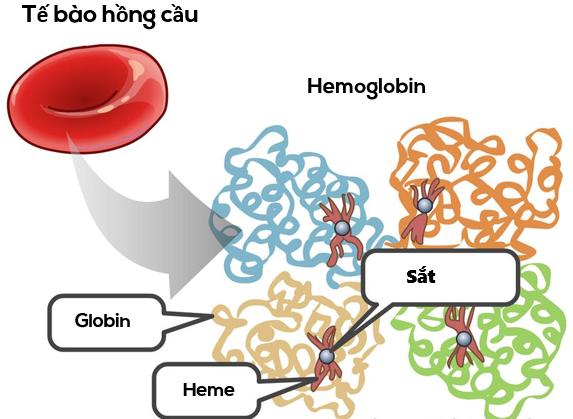
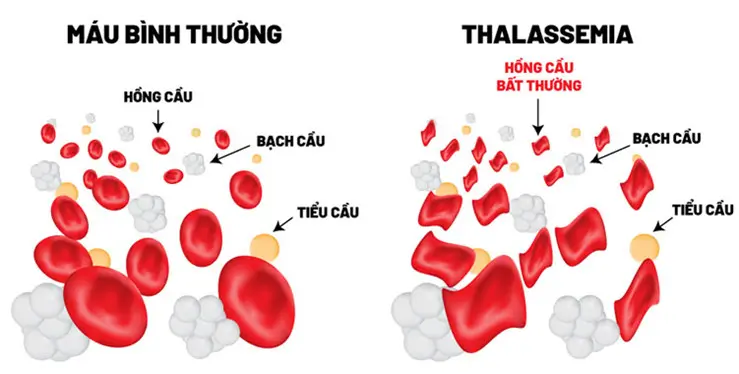
Beta thalassemia là một trong những bệnh di truyền lặn trên nhiễm sắc thể thường làm giảm khả năng sản xuất hemoglobin – một loại protein trong tế bào hồng cầu có chức năng mang oxy đi khắp các cơ quan trên cơ thể. Những người bị beta thalassemia sẽ có nồng độ hemoglobin trong máu thấp dẫn đến thiếu oxy trong nhiều phần cơ thể. Người mắc cũng thiếu máu (gọi là anemia) khiến da nhợt nhạt, mệt mỏi, suy nhược cơ thể và nhiều các biến chứng nguy hiểm khác. Những người bị beta thalassemia có nguy cơ phát triển các cục máu đông bất thường.
Xem thêm: Tìm hiểu về hemoglobin là gì
Phân loại
Beta thalassemia được chia làm 3 dạng dựa trên mức độ sản xuất beta globin bị suy giảm: thalassemia thể nặng (còn được gọi là thiếu máu Cooley), thalassemia thể trung gian và thể ẩn. Trong ba dạng này, thiếu máu Cooley biểu hiện trầm trọng hơn.
Những dấu hiệu và triệu chứng của thalassemia thể nặng xuất hiện trong 2 năm đầu đời. Trẻ nhỏ sẽ bị thiếu máu nặng nguy hiểm đến tính mạng. Chúng chậm tăng cân hoặc phát triển như đúng độ tuổi của mình, vàng da, vàng mắt. Ngoài ra, trẻ có thể có bị gan to, lách to, tim to và có thể không có xương, hoặc dậy thì muộn. Nhiều trường hợp bị thalassemia thể nặng có các triệu chứng trầm trọng như trên cần được truyền máu thường xuyên để làm đầy hệ thống cung cấp các tế bào hồng cầu. Lâu dần, truyền máu thường xuyên trong khoảng thời gian dài sẽ dẫn đến ứ sắt trong cơ thể gây ra các bệnh liên quan đến gan, tim, hormone.
Thalassemia thể trung gian thì nhẹ hơn thalassemia thể nặng. Các dấu hiệu hoặc triệu chứng của thalassemia thể trung gian có thể xuất hiện sớm hoặc muộn trong giai đoạn sống. Những người mắc bị thiếu máu nhẹ cho đến nặng có thể phát triển chậm và bất thường xương.
Người mắc bệnh thalassemia thể nhẹ sẽ có 1 gen bị đột biến và 1 gen bình thường. Bệnh nhân thường không có triệu chứng và bị thiếu máu nhẹ. Các tế bào hồng cầu có kích thước nhỏ bất thường, và có thể không đáp ứng với các chất bổ sung sắt. Bệnh có thể ảnh hưởng cho con cháu do di truyền.
Biểu hiện lâm sàng của bệnh Beta thalassemia
Triệu chứng của beta thalassemia sẽ tùy thuộc vào từng thể được di truyền. Với bệnh nhân mắc thể nhẹ thường không có triệu chứng đặc hiệu nào, người bệnh có thể gặp vấn đề về thiếu máu nhẹ.
Ở bệnh nhân mắc beta thalassemia thể nặng sẽ có những triệu chứng sớm trong những năm đầu đời bao gồm: da nhợt nhạt, hay cáu kỉnh, ăn không ngon miệng, dễ bị nhiễm trùng. Theo thời gian, nhiều triệu chứng sẽ xuất hiện, bao gồm: tăng trưởng kém, bụng lồi, da và mắt hơi vàng.
Với bệnh nhân mắc beta thalassemia thể trung gian, bệnh có thể gây ra các triệu chứng thiếu máu từ trung bình đến nặng bao gồm: cực kỳ mệt mỏi, da nhợt nhạt, tăng trưởng chậm, xương yếu, lá lách to.
Bệnh beta thalassemia có nguy hiểm không?
Phải khẳng định beta thalassemia là căn bệnh cực kỳ nguy hiểm bởi nó không chỉ gây tổn hại sức khỏe người bệnh, gây tàn phá cấu trúc trong cơ thể, ảnh hưởng đến giống nòi, đe dọa đến tính mạng mà nó còn tạo thêm gánh nặng cho gia đình, xã hội.
Beta thalassemia gây biến chứng vô cùng nguy hiểm. Nếu không điều trị, lá lách, gan và tim của người bệnh có xu hướng to ra dẫn đến việc có thể phải cắt bỏ lá lách trong trường hợp lách quá to và gây đau. Điều này cũng làm tăng nguy cơ nhiễm khuẩn khi bệnh nhân đã cắt bỏ lá lách.
Với bệnh nhân mắc bệnh ở thể trung gian và nặng sẽ gặp nhiều vấn đề về xương như xương trở nên mỏng, giòn và biến dạng. Người bệnh sẽ cần được truyền máu thường xuyên để duy trì cuộc sống. Tuy nhiên việc truyền máu tăng nguy cơ tích tụ sắt gây ảnh hưởng nghiêm trọng đến các cơ quan của cơ thể.
Nguyên nhân gây ra bệnh beta thalassemia
Đột biến trên gen HBB gây ra bệnh beta thalassemia. Gen thalassemia mang thông tin tổng hợp protein gọi là beta-globin, đơn vị cấu tạo nên hemoglobin. Hemoglobin có chứa 4 đơn vị cấu tạo, điển hình là 2 đơn vị cấu tạo của beta-globin và 2 đơn vị cấu tạo của protein khác gọi là alpha-globin. Một số đột biến trên gen HBB ngăn chặn sản xuất beta-globin.
Không có beta-globin gây ra thalassemia beta-zero. Những đột biến khác trên gen HBB cho phép beta-globin được tạo ra nhưng số lượng bị giảm gây ra thalassemia beta-plus. Tình trạng bị beta-zero thalassemia hay beta-plus thalassemia không cho biết mức độ nghiêm trọng của bệnh mà phụ thuộc vào việc chuẩn đoán người bệnh bị thalassemia thể nặng hay thể vừa.
Thiếu beta-globin dẫn đến giảm số lượng hemoglobin chức năng, không đủ số lượng hemoglobin khiến các tế bào máu phát triển không bình thường gây thiếu các tế bào hồng cầu trưởng thành. Việc thiếu các tế bào hồng cầu trưởng thành dẫn đến thiếu máu và các vấn đề sức khỏe liên quan đến beta thalassemia.
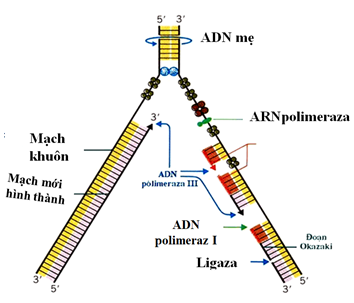
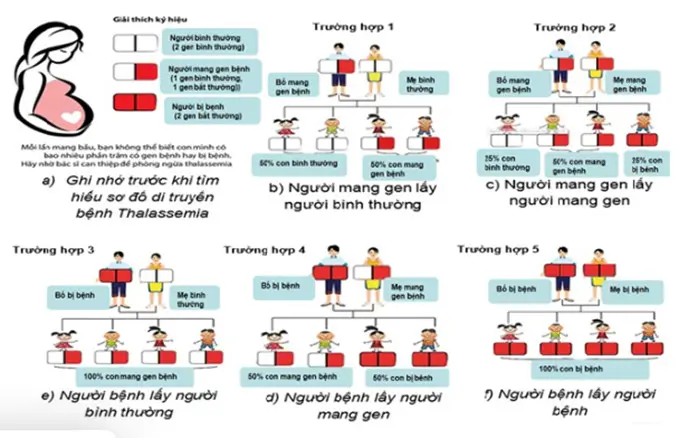
Cơ chế di truyền
Thalassemia bao gồm cả thể beta và alpha là một bệnh di truyền lặn, được di truyền từ bố mẹ cho các thế hệ sau. Theo quy luật di truyền, bố và mẹ mỗi người truyền một nửa vật chất di truyền cho con cái. Theo sơ đồ dưới đây, nếu một trong hai bố mẹ bình thường, người kia mang gen bệnh, tỷ lệ sinh con là 50% bình thường: 50% con mang gen bệnh. Nhưng nếu cả hai bố và mẹ đều mang gen bệnh thì tỷ lệ sinh con là 25% bình thường: 50% mang gen bệnh: 25% bị bệnh.
Xem thêm: Bệnh tan máu bẩm sinh thể Alpha (Alpha thalassemia) là gì?
Biện pháp phòng ngừa
Để phòng bệnh beta thalassemia hiệu quả, các cặp đôi nên thực hiện khám tiền sản, làm các xét nghiệm gen để có sự lựa chọn đúng đắn khi mang thai, giúp tránh việc sinh ra những đứa trẻ có nguy cơ cao là thể mang beta thalassemia.
Trong trường hợp khi xét nghiệm cặp đôi đều mắc gen bệnh, bác sĩ sẽ khuyến nghị thực hiện thụ tinh ống nghiệm kết hợp với sàng lọc phôi với phương pháp xét nghiệm di truyền tiền làm tổ (PGT) trước khi chuyển phôi. Việc sàng lọc phôi sẽ lựa chọn được những phôi khỏe mạnh không mang bệnh beta thalassemia. Những phôi khỏe mạnh sau khi sàng lọc sẽ được tiến hành chuyển vào buồng tử cung của người phụ nữ.
Cặp vợ chồng dự tính có con hoặc trong thời kỳ đầu của thai kỳ, nên thử máu để xác định xem mình có phải là người có gen bệnh này hay không. Nếu gia đình của một trong hai người có gốc từ những vùng như gốc Á, gốc Phi Châu, Trung Đông và vùng Địa Trung Hải; hoặc nếu gia đình họ có tiền sử bị bất cứ bệnh rối loạn máu hoặc thiếu máu nào. Xét nghiệm này rất cần thiết trong việc xác định xem có bất cứ nguy cơ nào về vấn đề em bé có bị ảnh hưởng bởi bệnh rối loạn máu di truyền hay không.
Nếu bạn là người có gen beta thalassaemia, những người khác trong gia đình cũng có thể có gen này và có nguy cơ có con bị bệnh beta thalassaemia dạng trầm trọng. Tất cả người khác trong gia đình và người phối ngẫu đều nên làm xét nghiệm ADN để biết tình trạng gen của mình trước khi có con.
Điều trị bệnh Beta thalassemia
Hiện nay, có hai phương pháp điều trị chính của bệnh beta thalassemia là truyền máu và thải sắt. Bên cạnh đó, một số biện pháp khác cũng được áp dụng trong điều trị thalassemia cho những trường hợp cụ thể.
Truyền máu: bệnh nhân mắc thalassemia tùy mức độ sẽ bị thiếu máu mạn tính và cần phải truyền máu định kỳ. Việc này có mục đích duy trì nồng độ huyết sắc tố trước truyền là 90 – 100g/l (đối với nhóm nặng, phụ thuộc truyền máu).
- Thải sắt: mục đích để chống quá tải sắt nhằm hạn chế biến chứng trên các tổ chức, cơ quan trong cơ thể. Bệnh nhân thường phải duy trì dùng thuốc thải sắt cả đời.
- Cắt lách: được chỉ định trong các trường hợp: khi bệnh nhân tăng nhu cầu truyền máu, lách to gây cản trở sinh hoạt hàng ngày của người bệnh hoặc gây đau, giảm bạch cầu hoặc tiểu cầu do cường lách.
- Ghép tế bào gốc: đây là phương pháp điều trị hiện đại và có thể chữa khỏi bệnh. Tuy nhiên khả năng có người cho phù hợp để cấy ghép là rất thấp và chi phí điều trị điều trị của phương pháp này cũng rất tốn kém.
- Bên cạnh đó, với những bệnh nhân đã bị nhiễm sắt nặng tại các bộ phận như gan, tim… thì tỷ lệ thành công sẽ thấp hơn.
- Chăm sóc toàn diện: là một trong những ưu tiên hàng đầu với bệnh nhân mắc beta thalassemia, việc chăm sóc toàn diện sẽ phòng ngừa và hạn chế các biến chứng của bệnh, giúp người bệnh có chất lượng cuộc sống tốt hơn.
- Điều trị biến chứng: tùy theo biểu hiện, việc điều trị biến chứng sẽ được điều trị dựa vào tình trạng bệnh nhân mắc phải như có suy tuyến nội tiết, bệnh nhân mắc đái tháo đường, suy tim, xơ gan, loãng xương, rối loạn đông máu…
Như vậy, bài viết đã cung cấp các thông tin về bệnh tan máu bẩm sinh thể beta. Hy vọng, qua bài viết này có thể cung cấp những thông tin hữu ích về bệnh beta thalassemia cũng như cách phòng ngừa đến mọi người. Để cập nhật thêm các thông tin, mời bạn tiếp tục theo dõi các bài viết trên website của ADNVIETNAM.
Nguồn tham khảo:
Beta Thalassemia